


Fernández Rodríguez
Research themes
Quantum chemistry: Accurate Evaluation of Molecular and Intermolecular Properties
Research
FORMER CIQUS RESEARCH GROUP, 2011-2015
Currently at the Quantum Chemistry Group (USC): http://qfminerva.usc.es
The theoretical description of intermolecular interactions is a challenge, not only because of the importance these interactions have in the interpretation of a large number of physical, chemical and biological problems; but also because a considerable number of previous theoretical studies failed, a failure that pointed out the need of improving the quality of the calculations in order to get accurate results. The high accuracy of the available experimental data also poses a challenge for the theoreticians.
Van der Waals and H-bond complexes are interesting systems, because of the weak interactions (dominated by dispersion) in the former, and the presence of the later in many key biological interactions.
From the theoretical point of view, the dispersion interactions are the most difficult to describe, as they require not only accurate methods like the coupled cluster, but also large and flexible basis sets. We are working on the following projects.
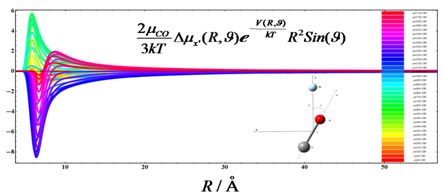
Van der Waals Complexes: Accurate Evaluation of Interaction Properties
The main objective of this project is to accurately evaluate van der Waals complex interaction properties.
Coupled cluster methods are especially suitable for the study of this type of systems, due to, among other properties, their size extensive character. In particular, the CCSD and the CCSD(T) methods together with large basis sets provide a correct description.
We extend the coupled cluster code within the DALTON program in order to reduce the scaling and to deal with larger complexes. For this we use the Cholesky decomposition of the integrals and the denominators. Response theory is used to evaluate the properties.
Theoretical study of molecular complexes of astrophysical interest
We carry out this project together with the group of Prof. J. Flores Rodríguez at the Univ. of Vigo. The aim is to develop and apply electronic structure quantum chemistry methods to the study of potential energy surfaces and intermolecular interactions, to the resolution of the rovibrational equations, and to the analysis of the evolution of energetically excited complexes.
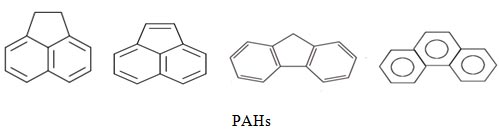
All the applications deal with molecular complexes of astrophysical interest, like those with polycyclic aromatic hydrocarbons (PAHs), and neutral or charged atoms or molecules.
The PAHs are, together with other molecules, responsible for the diffuse interstellar bands (DIBs), and for the unidentified infrared bands (UIRs). We analyze the electronic structure, both in the ground and in the principal excited states, and the rovibrational and dynamical behaviour of the complexes.
The results are compared to those available, obtained with high-resolution spectroscopic techniques or using dynamics. The conclusions are of great interest for modelling astrophysical processes, and for advancing the understanding of physical mechanisms of general interest.
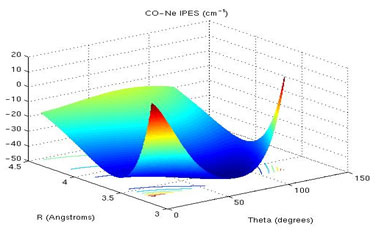
Accurate ab initio study of van der Waals and H-bonded complexes: Structure and reactivity
We deal with the study of intermolecular interactions in van der Waals and H-bonded complexes, by evaluating accurate intermolecular potential energy surfaces, the corresponding dynamics, and interaction properties using coupled cluster methods. Additionally, we use perturbation and density functional theory to compare our results.
Regarding the methodology, we carry out an extension of the available Cholesky-decomposed coupled cluster code within the DALTON program in order to be able to apply it in a more efficient way.
We also develop more efficient basis sets which provide accurate interaction properties with a smaller number of functions.
FORMER CIQUS RESEARCH GROUP, 2011-2015